醫療器材檢測相關專題
生醫產品上市前驗證程序 (含ISO 10993-1:2018 更新版說明)
撰稿人: 生醫驗證組 魏琪珍 博士
事實上安全並非等同於沒有風險,而是經整體風險利益評估 (Risk/Benefit ratio) 後,利益大於風險的結果。因此在醫療器材安全評估過程,如何有效使用風險管理一直是重要的課題,醫療器材製造商應在符合使用目的之同時,經由一系列風險分析、評估及管理,從產品的設計、訂定規格、研發、製造、驗證、確效,乃至臨床使用、上市後使用資訊等,於整個醫療器材生命周期實施風險管理 (如下圖一),盡可能透過各種預防手段減少風險,使患者利益高於風險,並告知使用者殘餘風險,始能符合各國法規要求,同時及早於產品發展階段發現問題,降低後期矯正所需支付的龐大成本。
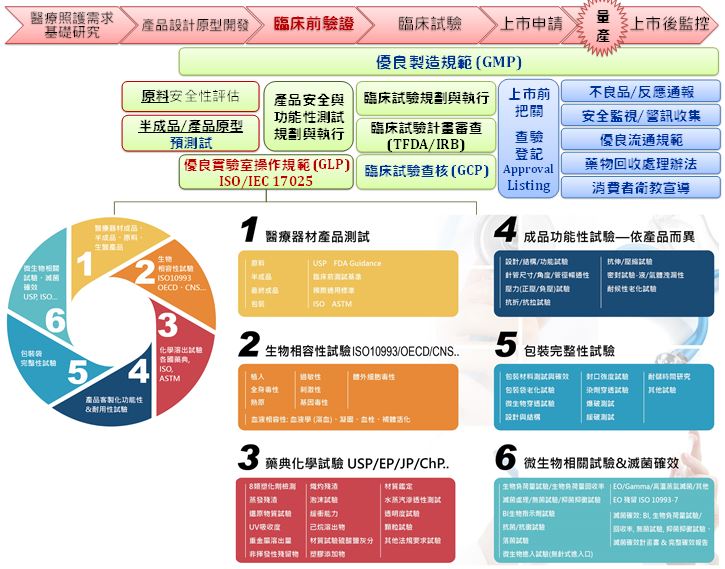
圖一、塑膠中心醫療器材檢測驗證能量 (全生命周期管理體系)
醫療器材廠商可以依據下圖二4項原則思考如何把關產品的安全:
1. 風險管理:以ISO 14971為基礎,在產品生命周期 (Total Product Life Cycle) 中,從各個階段 (產品設計、製造、上市) 的各種面向 (風險分析工具、醫療器材指引及標準、上市後資訊等) 蒐集風險資訊;使用ISO 14971 Annex C找出安全性可能受醫材特性影響的風險,評估醫療器材製造業在製造上常見之「可預見危害」例如: 操作危害、生物與化學危害、資訊危害或能量危害等,讓製造商能有效避免此危害之發生,降低醫療器材製造業之「製造失誤,避免因製造疏失產生人生命安全之問題。
2. 權責機關發佈指引:醫療器材涉及療效與生命安全習習相關,因此國內外權責機關對於醫療器材均有相關法令與規範予以嚴格管理,以確保上市使用的功能性與安全性。在此前提下,各國衛生主管機關均持續針對特定產品發行指引 (guidance),供做權責機關產品審查員、業者、民眾以及相關行業可遵循的一致性標準,如:我國醫療器材臨床前測試基準/技術基準、美國FDA guidance …。
3. 產品相關之基準、標準、規範:除了以風險管理概念全面考量產品安全評估為主,輔以針對特定醫療器材產品制定的標準,如:ASTM標準、ISO標準、CNS標準、各國藥典等國際、區域或國家標準、著名技術組織、相關科學書籍或期刊,其中可能包含產品物理性、化學性、電性安全、電磁相容性、生物相容性、機械功能性等各面向的要求,可補足風險管理未能全面涵蓋的內容。
4. 規格或宣稱特殊功能、療效:最後則是針對製造廠所提供的產品仿單內容:一般指型錄Catalog、使用說明書Instruction for use、操作手冊Operation manual等,其中包含宣稱產品用途、注意事項、型號規格、圖樣等。若宣稱產品具特殊性質或功效,則需提供相關資料佐證其描述,如:抗菌、抗血栓形成、可分解性…等。
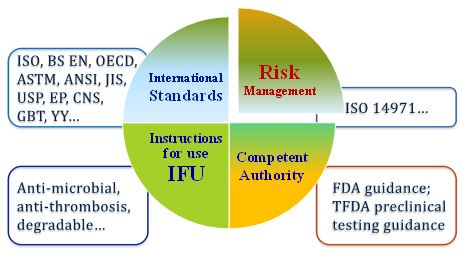
圖二、醫療器材產品安全性評估準則
上述完整的醫療器材安全性評估幾乎全面性涵蓋以下範疇:產品規格、電性安全、電磁相容性、生物相容性、功能性、機械物理性、化學性、無菌性等試驗的執行。其中,由於生物相容性牽涉到昂貴的收費、較長的試驗期間與動物試驗的執行,因此在進行前就必須更嚴密衡量試驗的需要性與合適性。依據ISO 10993的規範,醫療器材最終成品的生物相容性不僅取決於使用物質,還包括物質處理、製造方法 (含滅菌過程)、以及可能出現的製程殘餘物而定。選擇適合醫療器材生物性評估的試驗項目時,應依據器材物質的化學特徵,以及人體暴露性質、程度、頻率和時間而定。委託者於評估生物相容性時,應考量是否有必要進行每一項建議試驗,若未依此進行測試項目時,應檢附相關文獻或科學性評估報告,以證實產品仍具有相等之安全及功能。
2018年10月發行的ISO 10993-1最新版本對於生物性評估原則有確切的規範,其主要變更如下,塑膠中心已將新版要求納入委託試驗物質的生物性評估規劃中,歡迎有興趣深入了解的廠商與我們進一步聯繫。
1. 附錄A (Annex A)
(1) 2018年版新增下列6欄位
A. 物理化學資訊 (Physical and/or chemical information)
B. 原料致熱性(Material mediated pyrogenicity)
C. 慢性毒性 (Chronic toxicity)
D. 致癌性 (Carcinogenicity)
E. 生殖/發育毒性 (Reproductive/developmental toxicity)
F. 降解性 (Degradation)
(2) 以亞急性、亞慢性毒性2個單獨欄位取代2009年版的單一欄位 「亞慢性 (亞急性)毒性」
(3) 以試驗評估終點/指標 (E, Endpoints) 取代原2009年版的附錄A中顯示的試驗 (X, Tests)
(4) 各分類醫療器材建議執行試驗評估內容新舊版間出現差異,以接觸體表、黏膜、B類產品 (接觸時間介於24小時至30天內) 為例,有如下表的差異:
| 建議執行項目 | 2018年版 | 2009年版 |
| 物理化學資訊 |
ü
|
|
| 體外細胞毒性 | ü | ü |
| 敏感性 | ü | ü |
| 刺激性 | ü | ü |
| 急性毒性 | ü | |
| 亞急性毒性 | ü | |
| 植入性 | ü |
| 2018年版 | 2009年版 |
|
以風險管理程序實施生物性評估指引
“Guidance on the conduct of biological evaluation within a risk management process” |
風險管理程序指引 “Guidance on the risk management process” |
4. 新增評估非接觸性醫療器材 (Non-contacting medical devices) 與短暫接觸醫療器材 (Transitory-contacting medical device) 的資訊。
5. 新增評估奈米原料 (nanomaterials) 與吸收性物質 (absorbable materials) 的資訊。
6. 新增吸入性氣體管路醫療器材 (Biocompatibility evaluation of breathing gas pathways in healthcare applications) 生物相容性評估參考ISO 18562 (所有部分) 舉例說明。
ISO10993生物相容性試驗建議項目包括:體外細胞毒性試驗 (萃取法、直接接觸法、間接接觸法)、刺激性試驗 (皮膚、皮下、眼睛、口腔、陰道、腸道…)、過敏反應試驗 (極大化法、封閉式貼片法、小鼠淋巴結法)、毒性試驗 (急性、亞急性、亞慢性和慢性)、血液相容性試驗 (致血栓、凝固、血小板、血液學及免疫性: 補體活性)、植入性試驗 (骨、肌肉、皮下)、基因毒性試驗 (體外、體內試驗)、致癌性試驗、生殖和發育效應試驗以及免疫毒性試驗。
對於風險等級較低的醫療器材最基本且常被選用於分析其生物相容性的試驗為:體外細胞毒性試驗、刺激性試驗與過敏反應試驗三項;至於驗證醫療器材等級的原料安全性評估,則可參照美國藥典USP 88要求,執行急性毒性試驗、皮內刺激試驗與肌肉植入試驗,後續更可依據各國的要求,進行原料藥主檔案 (Drug Master File, DMF) 的申請登記,為原料創造更高價值與增加應用層面。
為達試驗結果的適用性、真實性與可靠性,醫療器材廠商必須考量下列重點,才能確保試驗規劃與執行結果符合原預期的驗證目的:
1. 實驗室資格確認:不論透過那些生物相容性試驗,以確保醫療器材不致於在使用時直接或透過釋出物質成分對人體產生局部或全身不良反應、具致癌性或產生生殖和發育不良反應; 其中最重要的是確保相關試驗能被正確執行並獲得精確及可信的結果,才能為醫材產品的安全性嚴格把關。財團法人塑膠中心驗證實驗室具備完善的試驗設施及動物飼育環境,以及優質的試驗經驗與管理品質要求,確保試驗的準確與客觀性,在實驗室嚴謹的管理制度下,維持試驗設備的完備與精確、試驗人員定期接受技術訓練考核,藉此提供具公信力及高可信度的測試報告與高標準的試驗品質。
2. 試驗方法選用:由上述內容可知,同樣是體外細胞毒性試驗、刺激試驗、植入試驗或毒性試驗,因產品種類與特性的差異,往往需更精準的判斷後,才能選用最適合的試驗方法證實其生物相容性;透過與委測實驗室的討論與溝通,加上實驗室累積的各種執行經驗,都有助於做出最正確的判斷,省卻不必要的浪費。
3. 試驗條件確認:通常試驗的進行即使有國際標準可供參考,其中都不必然只有一種試驗條件,以ISO 10993-12為例,其中試驗物質萃取比例至少4種以上、萃取條件也至少4種,加上萃取溶液的選項更多,若無法事先確立試驗進行的所有條件,將導致結果的錯誤,而無法如預期證實產品的安全性。
4. 執行方法確認:試驗進行完畢後,如何評估試驗體系確實如預期正確運作,則為委託者另一項功課,例如透過陰性、陽性試驗對照組的結果,即可確認試驗的可接受度。
5. 結果/結論判讀:標準方法不一定均包含結果判定基準,或者即使標準內容明訂判定基準也不必然適用於送測產品,因此如何依照產品本身的風險程度訂定合適的允收標準,也是產品上市前驗證程序中需確認的一項重要課題。塑膠中心驗證實驗室多項試驗均已取得TAF ISO/IEC 17025認證,包含醫療器材成品、半成品、原料之生物相容性、無菌試驗與包裝袋結構完整性試驗 (加速熱老化試驗、封口強度試驗、爆破試驗、緩破試驗及染劑穿透試驗);為力求試驗品質更上層樓,更於107年2月13日通過OECD GLP (經濟合作暨發展組織優良實驗室操作規範) 符合性登錄,國內生醫領域業者之醫療產品/醫材原料/半成品,將可在本中心符合OECD GLP規範的品質要求下,提供廣為國際各國認可之生物相容性試驗驗證服務,包括ISO 10993體外細胞毒性試驗、各部位刺激試驗、過敏試驗、長短期毒性試驗、血液相容性試驗、植入試驗、熱原試驗等,其餘生物相容性試驗則仍持續建立中;此外,依照各產品特性執行滅菌確效、微生物相關試驗、化學溶出試驗與客製化產品功能性評估等臨床前試驗,以提供醫療器材廠商整體的產品安全性試驗規劃等服務。
未來,醫藥高分子材料將成為第一大原材料項目與應用趨勢,塑膠中心將跨入高值化高分子醫材應用範疇,先後陸續強化相關硬體設備,包含即將於2018年興建落成的「高分子醫材大樓」。高分子醫材大樓立足中部,為補足高分子醫材關鍵技術缺口,與北部生醫光電及南部金屬醫材相呼應,成為發展高分子醫材產業的重要優勢。高分子材料需具備與人體的器官、組織細胞及生物大分子相容,並對人體無毒、無熱原、不致癌等特性,因此本實驗室將已陸續提供亞急、亞慢性及慢性毒性測試、植入試驗;並建置基因毒性試驗等服務,以建立符合多項國際法規與全套生物相容性試驗能量,達成塑膠中心扶植醫療器材產業與創造客戶價值之理念與願景!
【聯絡窗口】
| 醫療器材檢測驗證 | |
諮詢專線:(04)23595900 #638 & #643 信箱:md3d@pidc.org.tw LINE 帳號:pidcmd3d  |