國際醫療器材管理法規發展現況—臨床評估報告要求
前言
近年來台灣醫療器材產業外銷出口比例大幅提升,但是醫療器材產品在上市前皆須向欲銷售國家之衛生主管機關提交上市前申請,而各國之衛生主管機關皆會依據其國家法規或國際標準內容,要求廠商提交相關申請資料,以證明產品之安全性與功效性皆符合法規、標準、預期用途…等要求;不論是美國510(k)或是歐盟CE Mark皆逐漸開始注重產品臨床評估相關資料,此部分可以類似品比對、文獻蒐集、實質進行臨床試驗等方式證實產品之安全性與功效性。
臨床評估(Clinical Evaluation)
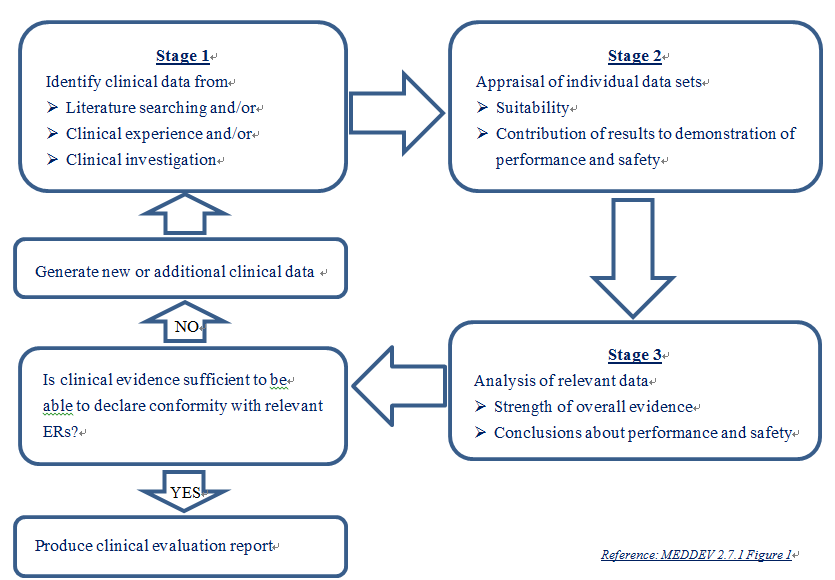
歐盟衛生主管機關最近開始要求驗證單位(Notified Body)須依據MEDDEV 2.7.1要求從嚴審查臨床評估報告內容,所以先前已取得CE Mark產品之臨床評估報告內容已不符合驗證單位要求,近來常被退件或要求提供更多資料以佐證產品安全性與功效性。MEDDEV 2.7.1建議蒐集臨床數據(Clinical Data)方式有三種: (1)臨床文獻、和/或(2)臨床經驗(類似品比對)、和/或(3)臨床試驗,並對蒐集到之臨床數據逐一評估其適用性及數據有效性,確認臨床證據是否符合產品基本要求(ESR);同時,須將評估人員之名單、資格及學經歷說明於臨床評估報告中,以證明評估人員具備產品技術及應用的能力、研究方法設計及分析能力。臨床評估報告之內容須與風險管理報告、標籤與使用說明書之敘述一致,而臨床評估報告之結論須證實臨床數據符合基本要求(ESR)、證明產品之安全性與功效性符合要求並確認產品之效益大於風險。
臨床評估報告須依據MEDDEV 2.7.1 附件E之架構內容撰寫,如下:
1. General Details
2. Description of the device and its intended application
3. Intended therapeutic and/or diagnostic indications and claims
4. Context of evaluation and choice of clinical data types
5. Summary of the clinical data and appraisal
6. Data Analysis
7. Conclusion
近年來台灣醫療器材產業外銷出口比例大幅提升,但是醫療器材產品在上市前皆須向欲銷售國家之衛生主管機關提交上市前申請,而各國之衛生主管機關皆會依據其國家法規或國際標準內容,要求廠商提交相關申請資料,以證明產品之安全性與功效性皆符合法規、標準、預期用途…等要求;不論是美國510(k)或是歐盟CE Mark皆逐漸開始注重產品臨床評估相關資料,此部分可以類似品比對、文獻蒐集、實質進行臨床試驗等方式證實產品之安全性與功效性。
臨床評估(Clinical Evaluation)
歐盟衛生主管機關最近開始要求驗證單位(Notified Body)須依據MEDDEV 2.7.1要求從嚴審查臨床評估報告內容,所以先前已取得CE Mark產品之臨床評估報告內容已不符合驗證單位要求,近來常被退件或要求提供更多資料以佐證產品安全性與功效性。MEDDEV 2.7.1建議蒐集臨床數據(Clinical Data)方式有三種: (1)臨床文獻、和/或(2)臨床經驗(類似品比對)、和/或(3)臨床試驗,並對蒐集到之臨床數據逐一評估其適用性及數據有效性,確認臨床證據是否符合產品基本要求(ESR);同時,須將評估人員之名單、資格及學經歷說明於臨床評估報告中,以證明評估人員具備產品技術及應用的能力、研究方法設計及分析能力。臨床評估報告之內容須與風險管理報告、標籤與使用說明書之敘述一致,而臨床評估報告之結論須證實臨床數據符合基本要求(ESR)、證明產品之安全性與功效性符合要求並確認產品之效益大於風險。
臨床評估報告須依據MEDDEV 2.7.1 附件E之架構內容撰寫,如下:
1. General Details
2. Description of the device and its intended application
3. Intended therapeutic and/or diagnostic indications and claims
4. Context of evaluation and choice of clinical data types
5. Summary of the clinical data and appraisal
6. Data Analysis
7. Conclusion