產品品質是由製造過程所決定
產品品質是由製造過程所決定

FDA管轄的產品包含食品、藥物、生物製劑及醫療器材,其遵循之品質系統,又稱為現行優良製造規範(Current Good Manufacturing Practice, cGMP),其中針對醫療器材的要求明訂於《美國聯邦法規法典》第21冊第820部分(21 CFR 820)—品質系統法規(Quality System Regulation, QSR)。
因此,進入美國市場的廠商在製造/ 販售醫療器材之前,必須按照QSR之要求建立品質管理系統。QSR為自我符合性,廠商在製造、販售醫療器材之前,應自行符合所適用的QSR規範;FDA不會核發QSR證書或認證。
QSR只提供一個框架,讓不同的醫療器材製造商,依據該器材當前發展狀況,更彈性地去符合品質系統的要求。每一製造商有其責任,針對每一類型或族群的醫療器材建立規範,以確保產品的安全及有效,並且訂定產品設計、製造、分銷、或其他活動的方法與流程,以符合品質系統之規範。FDA已於QSR法規中闡明品質系統所需的項目,且並未指示建立這些項目之特定途徑,因為QSR已經涵蓋了大部分的醫療器材及其生產要求,這樣的方式也讓製造商得以決定品質要項的詳細需求和範圍,並因應特殊的流程和產品來發展和執行特定的品質程序。
FDA已於2024年1月31日公布「《品質管理系統法規最終規則》(Quality Management System Regulation(QMSR)Final Rule),主要將QSR與ISO 13485:2016進行調和,並改稱為QMSR,預計於2026年2月2日正式生效,在此之前,醫療器材製造商仍應遵守現行 QSR 品質系統規定。
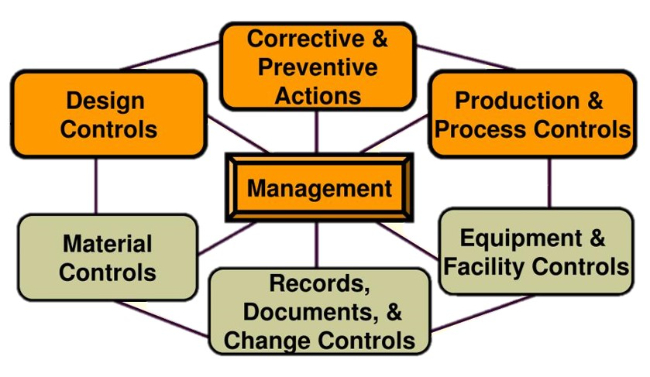
QSR系統有七大部分:Management, Design controls, Production & process controls, Records, Records/documents/change controls, Material controls, Facility & equipment controls, Corrective & Preventive actions.
其重點與主軸在於以下四個部分:
◎ Management
◎ Design controls
◎ Production & process controls
◎ Corrective & Preventive actions
其中,Corrective & Preventive actions又延伸出下列規範:
・Medical Device Reporting (MDR) regulation (21 CFR Part 803)
・Medical Device Tracking regulation (21 CFR Part 821)
・Corrections and Removals regulation (21 CFR Part 806)
【中低風險產品】
產品進行上市前通知申請(PMN=510(k))或產品列名(Listing)時,FDA對於製造廠的QSR符合性不會進行查核,待產品開始於美國販售時,才有可能被稽核。
【高風險產品(上市前核可(Premarket Application, PMA)/Class III 510(k)】
上市前核可之前,就會針對製造廠品質系統進行查核。其主要目的為:確認申請時所提交的生產相關資訊之正確性、評估廠商符合品質系統規範之能力。
| 醫療產業管理系統輔導 | |
| 劉組長 (04)23595900 #303 andy628@pidc.org.tw 0982-920-303 |
|
| 謝顧問 (04)23595900 #311 hanyu3516@pidc.org.tw 0982-920-297 |
洪顧問 (04)23595900 #685 aimeehung818@pidc.org.tw 0910-176-181 |