醫療器材製造業者的入門票
【輔導】醫療器材品質管理系統輔導
中華民國|國產醫療器材QMS登錄輔導
TFDA|醫療器材品質管理系統準則(QMS)
什麼是QMS?
醫療器材品質管理系統準則(簡稱: QMS),目的為審查醫療器材製造業者之品質系統,是否符合「醫療器材品質管理系統準則」(QMS),當貴公司通過嚴格的「醫療器材QMS檢查」後會核發醫療器材製造許可(效期三年),代表貴公司符合醫療器材法規要求,確保其達到產品的安全性、品質穩定性,及紀錄完整性等基本要求。
TFDA於2021.04.14公告「醫療器材品質管理系統準則」(QMS)並於2021.05.01跟隨母法「醫療器材管理法」一同生效,該準則共七章,總共79條。 廠商於2021年4月底前可申請GMP申請案(新案/後續案),而提出的GMP申請案其核可後的製造許可效期至多至113年4月30日(2024.4.30)。自2021.05.01起,新申請案應符合QMS準則並提出QMS申請,既有醫療器材製造業者(後續案)應符合QMS準則並提出QMS申請,至遲應於113年4月30 (2024.4.30)全面符合並取得醫療器材製造許可。
誰需要申請QMS?
若您是醫療器材製造業者,不論產品預計外銷或內銷,皆需要建立醫療器材品質管理系統並符合「醫療器材品質管理系統準則」,並報中央主管機關檢查合格取得製造許可後,始得製造。但部分經中央主管機關公告之品項,免取得製造許可。
【醫療器材製造業者】:依「醫療器材管理法」第10條
醫療器材製造業者,指下列二類業者:
一、從事醫療器材製造、包裝、貼標、滅菌或最終驗放。
二、從事醫療器材設計,並以其名義於市場流通。
【無需申請QMS之品項】:請參照「免取得醫療器材製造許可品項」
[TFDA於110年7月16日公告訂定「免取得醫療器材製造許可品項」(衛授食字第1101104548號)。]
ISO 13485與台灣GMP/QMS的發展關聯
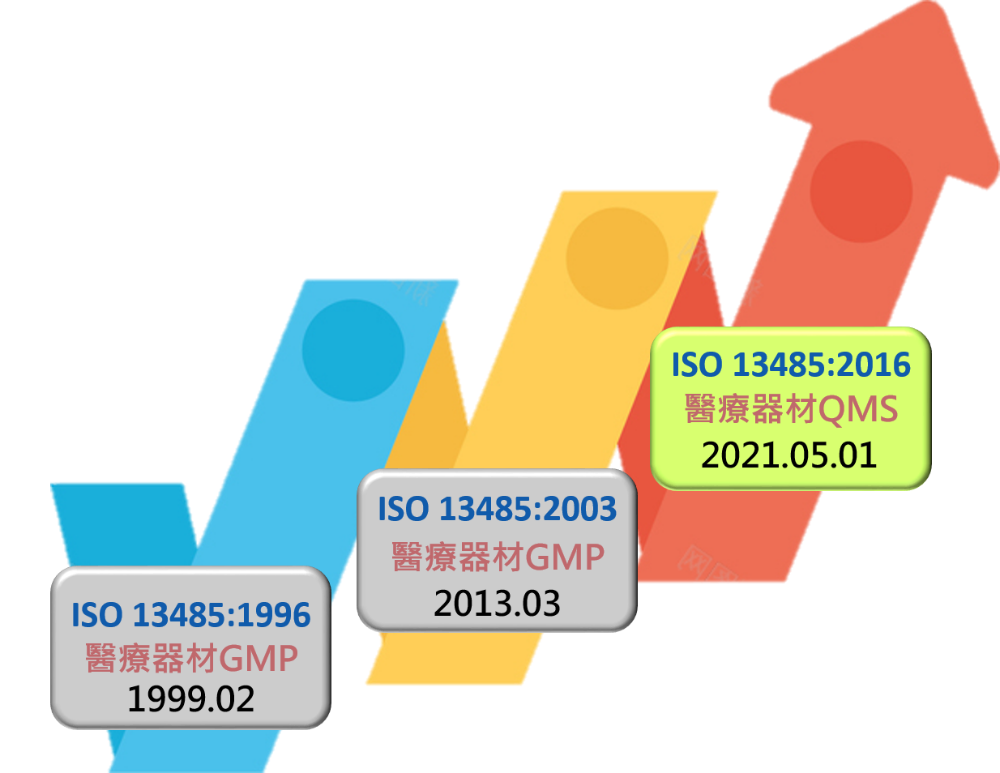
國產醫療器材製造業者申請QMS需要準備哪些資料 ? 國產醫療器材製造業者申請QMS應填具申請書,並備齊「醫療器材品質管理系統檢查及製造許可核發辦法」附表一所定文件、資料,並繳納費用後,向中央主管機關提出。
QMS對委託製造之檢附文件要求為何?
委託製造須於申請QMS所附資料中的產品製造流程圖呈現,並於流程圖中註明受託製造廠的名稱以及負責之活動階段。另外,流程具委託製造者,須檢附受託製造廠之QMS製造許可 (QMS認可登錄函)。
QMS相較於GMP之重要變革為何 ?
醫療器材品質管理系統準則(QMS)係參照ISO 13485:2016國際標準訂定,部分重要變革包括新增:
◎ 醫療器材檔案(Medical device file)
◎ 設計開發移轉(Design and development transfer)
◎ 設計開發檔案(Design and development files)
◎ 申訴處理(Complaint handling)
◎ 通報、回收及通告發布(Reporting to regulatory authorities)
...等以上新條文,並且強調醫療器材製造廠需要求應基於風險管理方法,管控品質管理系統所需的適當流程。同時對於「確效(validation)」、「查證(verification)」與「設計開發(Design and development)」等作業亦增加更明確之具體作業要求。
QMS申請案以「精要模式」申請者,其稽查重點為何?
QMS申請案「精要模式」以醫療器材品質管理系統準則第78條附表所列申請品項者為限。QMS申請案「精要模式」稽查要求大致與「標準申請模式」要求一致,惟稽查時將針對醫療器材品質管理系統準則 第11條(醫療器材檔案)、12條(文件管制)、13條(紀錄管制)、47條(生產與服務提供之管制)、55條 (追溯性之概述)、63條(申訴處理)、64條(通報、回收及通告發布)、69條(產品之監管與量測)、76條(矯正措施)、77條(預防措施實施檢查)。
【聯絡窗口】
| 醫療產業管理系統輔導 | |
| 劉組長 (04)23595900 #303 andy628@pidc.org.tw 0982-920-303 |
|
| 謝顧問 (04)23595900 #311 hanyu3516@pidc.org.tw 0982-920-297 |
洪顧問 (04)23595900 #685 aimeehung818@pidc.org.tw 0910-176-181 |