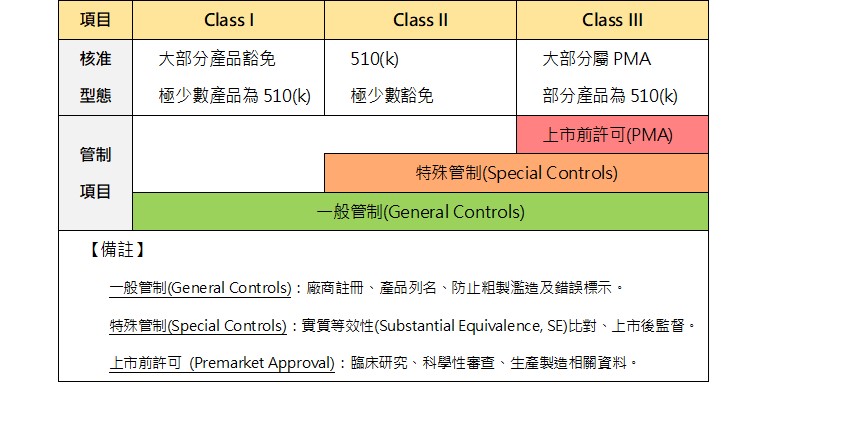
若任何人或製造商要將醫療器材產品(Class I, II, III)行銷至美國,除了部分豁免510(k)之品項及無須進行上市前核准(Premarket Approval, PMA)之品項外,皆必須在進口美國至少90天前向美國FDA提出上市前通知(Premarket Notification, PMN)申請,取得輸入許可(510(k) Clearance Letter)。
510(k)是向FDA提出的上市前申請文件,目的為證明申請510(k)的醫療器材不需進行上市前核准(PMA),且與已合法上市之產品具有相同安全性及有效性,此即實質相等性(substantially equivalent)。申請人或公司必須將欲申請上市的醫療器材與已經在美國FDA合法上市的一種或多種相似產品做比對,證明其具有實質相等性。
合法上市之器材的定義為在1976年5月28日之前合法上市的器材(Preamendment device)、或者從第III類中重新分類至II或I類的器材、或者通過510(k)申請程序證明具實質相等性的器材、或者依III類醫療器材評估定義的器材。這些醫療器材被稱為「Predicate Device(s)」。申請者必須提出描述性的資料,必要的時候,要提出功能性報告來證明與Predicate Device的實質相等性。
所以510(k)的資料是顯示比對性的資料,即待上市器材與Predicate Device的實質相等性。綜合以上內容可知,絕大部分產品在完成企業註冊、產品列名與GMP品質系統、完成510(k)申請後,即可上市。