
【輔導】醫療器材產品上市許可輔導
歐盟|CE Marking (MDR/ IVDR)申請輔導
CE Marking 歐盟醫療器材法規
歐盟是國內醫療器材外銷主要的市場之一,產品銷往歐盟須符合相關法規要求,並取得 CE 驗證。CE標誌為歐盟強制性驗證標誌,被視為製造商進入歐洲市場的護照。凡是貼有「CE」標誌的產品皆能夠在歐盟各成員國內銷售,無須符合每個成員國的要求,從而實現了商品在歐盟成員國範圍內的自由流通。
歐盟醫療器材法規
(Medical Device Regulation (EU)2017/745,簡稱EU MDR)
(Medical Device Regulation (EU)2017/745,簡稱EU MDR)
基於2011年爆發的PIP事件事件,歐盟開始重新審視醫療器材的管理方式。歐盟新醫療器材法(MDR)和新的體外診斷醫材法(IVDR)幾乎同步在2012年被提出。這些法規相較於過去的MDD、AIMD及IVDD有很大的改變。
最明顯的是,在於「直接生效性」。屬於Regulation的MDR和IVDR是直接生效;相對的,屬於Directive的MDD、AIMD及IVDD在歐盟立法後尚須經由各國在國內通過形成國內法才能生效。
歐盟為了使更安全的醫材在市面上流通,於是促進這些新醫療器材相關法規的產生,像是資料庫、追蹤系統、臨床評估、上市後監督 各方面都有顯著改變。
MDR於2017年5月5日正式發佈,並於2017年5月26日正式生效。歐盟委員會於2020年4月17日通過關於MDR實施日期推遲一年的建議,MDR生效日期推遲至2021年5月26日。
MDR關鍵時程
• 2017年5月26至2020年5月26日 – 3年過渡期(transition period)。
• 2021年5月26日 – MDR 正式實行日。
• 2022年5月27日 – MDD附錄IV證書失效。
• 2024年5月27日– MDD證書全面失效;MDD下是一類自我聲明,但在MDR下須透過驗證單位(NB)進行符合性評鑑的器材其在2020年5月26日之前已簽署的符合性聲明全面失效。
• 2025年5月27日– 所有MDD產品下架。
-歐盟市面上所有MDD/AIMDD證書相關產品必須撤下。
• 2017年5月26日 – MDR生效日。
• 2017年5月26至2020年5月26日 – 3年過渡期(transition period)。
• 2021年5月26日 – MDR 正式實行日。
• 2022年5月27日 – MDD附錄IV證書失效。
• 2024年5月27日– MDD證書全面失效;MDD下是一類自我聲明,但在MDR下須透過驗證單位(NB)進行符合性評鑑的器材其在2020年5月26日之前已簽署的符合性聲明全面失效。
• 2025年5月27日– 所有MDD產品下架。
-歐盟市面上所有MDD/AIMDD證書相關產品必須撤下。
MDR延期實施
MDR延期實施期限:
• Legacy Class III & IIb 植入式:2027 年 12 月
• Legacy Class IIb &Class IIa & Class Is/Im :2028 年 12 月
MDR過渡期延期需要滿足的條件 :
1.器材必須繼續符合指令90/385/EEC或指令93/42/EEC;
2.器材在設計和預期用途上沒有發生重大變化(關於重大變化的說明參見MDCG2020-3);
3.器材不會對患者、使用者或其他人的健康或安全,或對保護公眾健康的其他方面造成不可接受的風險;
4.2024年5月26日前,製造商已根據MDR第10(9)條建立了品質管理系統;
5.2024年5月26日前,製造商或其授權代表已根據MDR附錄VII第4.3節的規定,就對MDD指令的證書或符合性聲明所涵蓋的“Legacy device”向公告機構提交符合性評鑑的正式申請,要求進行符合性評鑑,並於2024年9月26日前,公告機構和製造商已按照MDR附錄VII第4.3節簽署認證書面協定。
管理模式
先前在歐盟地區有3個主要的醫療器材指令,分別為90/385/EEC (AIMDD)、93/42/EEC (MDD)、98/79/EC (IVDD),但於2017年5月正式以2017/745 (MDR)與2017/746 (IVDR)正式取代原先之指令,其中MDR具有4年轉換期(2021年取代原先之AIMDD、MDD);IVDR則具有5年轉換期(2022年取代原先之IVDD)。
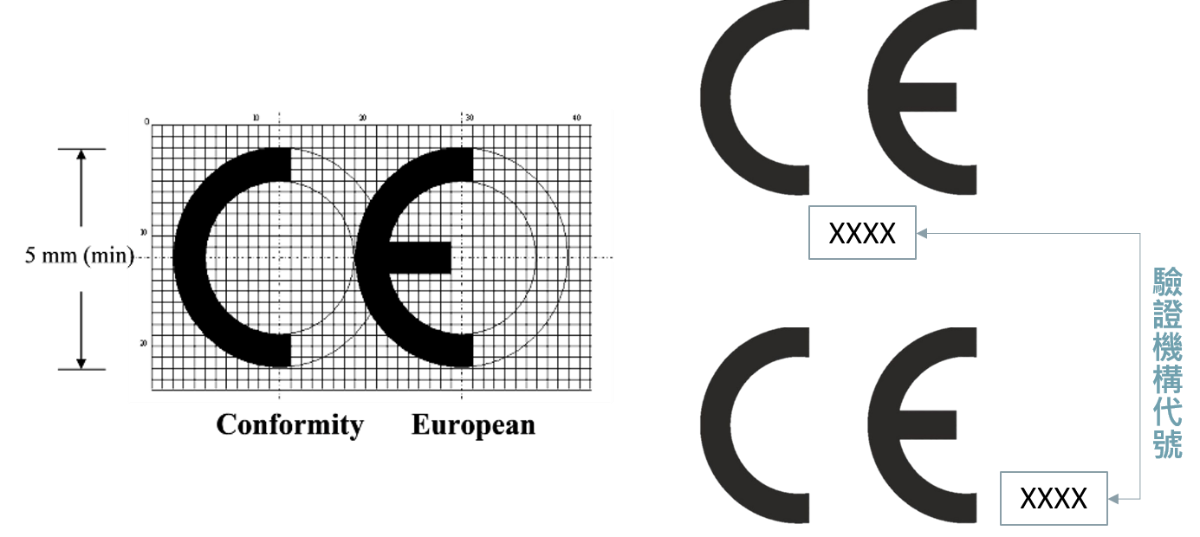
在這些法規中對於產品的分類、上市途徑有很詳細的敘述;而且在歐盟其醫療器材產品的分類準則是依接觸時間、是否造成創傷、適用位置及能量來源方式將產品進行分類,由申請者針對其器材特性選擇產品上市途徑,並連同產品技術文件 (Technical Construction File)向已獲得歐盟委員會批准的CE驗證單位(NB)提出驗證申請,通過驗證後即可將其產品貼上帶有該驗證單位代碼之CE標籤,才可將醫療器材產品在歐盟地區上市販售。
針對不同的經濟營運商(Economic Operators, EO)角色,若您是製造商(Manufacturer,MFR),於目前法規監管制度下, MFR承擔將產品銷售於歐洲市場CE Mark標誌的主要責任。目前,MFR需要因應法規變動採取行動並依據新法規要求取得CE Mark標誌之產品。
如果貴公司是醫療器材製造商,那麼您必須在2021年5月25日之前遵守MDR。建議貴公司盡快開始進行MDR之轉版。因為市場上眾多的醫療器材必須依照新的法規要求取得CE Mark產品證書,可預期的是驗證單位(NB)以及廠商都可能遇到瓶頸,且在過渡期間預期對驗證單位將產生一定壓力,早期啟動尤其重要。 建議盡快與您選擇之驗證單位(NB)進行聯繫及溝通,了解確認他們是否規劃根據新規定要求申請成為歐盟核可的驗證單位(NB),包括發證涵蓋之範圍,以及他們認為何時可取得相關資格。
歐盟要求位於歐盟境外製造商必須在歐盟境內指定一個歐盟授權代表 (European Authorized Representative),確保產品進入歐盟市場後的安全性與功效性。歐盟授權代表是指由位於歐洲經濟區EEA(包括EU與EFTA)境外的製造商明確指定的一個自然人或法人。該自然人或法人可代表EEA境外的製造商履行歐盟相關的指令和法律對該製造商所要求的特定的職責。
歐盟醫療器材上市流程
(01) 醫療器材之範圍與定義
(02) 風險分級 & 分類規則
(03) 選擇符合性評鑑程序
(04) 通用安全與性能要求(GSPRs)
(05) 調和標準 & 通用規則
(06) 技術文件 & 品質系統
(07) 註冊:組織角色、醫療器材、UDI
(08) 驗證機構以及相關單位審查
(09) DoC(符合性聲明)以及CE Marking
(10) 上市後管理
【聯絡窗口】
| 醫療產業管理系統輔導 | |
| 劉組長 (04)23595900 #303 andy628@pidc.org.tw 0982-920-303 |
|
| 范顧問 (04)23595900 #325 mandyfan@pidc.org.tw 0978-110-207 |
陳顧問 (04)23595900 #333 lilychen0703@pidc.org.tw 0919-602-859 |