原料可依據美國藥典USP<87>執行體外細胞毒性試驗、USP<88>執行三類生物相容性試驗 (皮內刺激試驗、急性系統毒性試驗、肌肉植入試驗),以確認其符合之醫療器材等級規格,共可分為Class I ~ Class VI;另外,可透過USP<661>等規範同時分析其溶出物。
USP<88>
亦可考量申請登錄Master File,可準備下列建議資料進行申請。
Master Files are regulatory dossiers that may contain information such as
-- Description of facilities
-- Manufacturing procedures and processes
-- Product formulation
-- Quality control procedures
-- Product characterization data
可申請登錄Master File 衛生主管機構如:
-- US FDA DRUG (Device) MASTER FILES
-- European Medical Agency (EMA)
-- Japanese Pharmaceutical and Medical Devices Agency (PMDA)
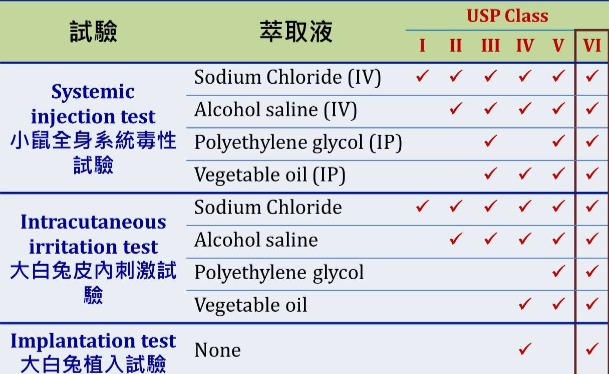 USP<88>
USP<88>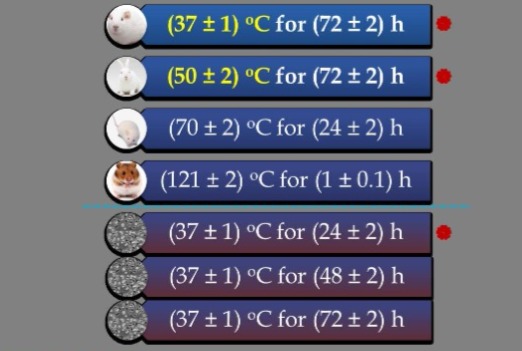 ISO 10993-12
ISO 10993-12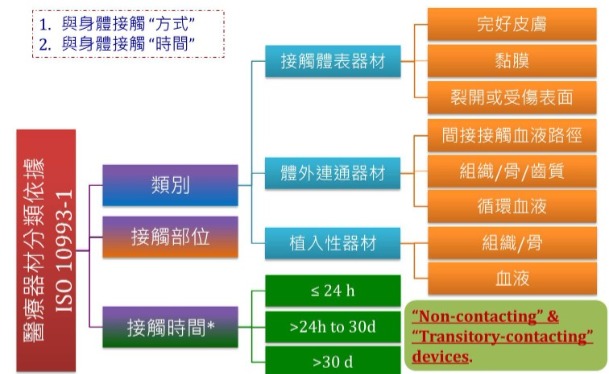 ISO 10993-1: 2018
ISO 10993-1: 2018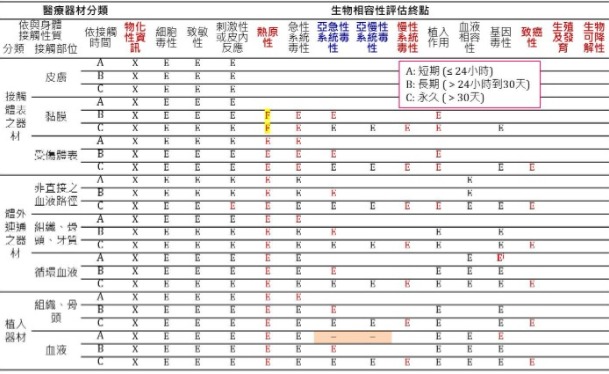 ISO 10993-1: 2018 Table A1
ISO 10993-1: 2018 Table A1